mag3D Figure1 Figure2 Figure3 PRESENTATION SYMPOSIUM FRANCO FINLANDAIS SYMPOSIUM Problème des phases The phase problem DVD Semi-conducteur organométallique organosilicié TiO2-rutile SiO2-stishovite Cu2O-cuprite K2PtCl6 Be-metal bases and 3D views Bases et vues 3D Si-et-Ge CaF2-fluorite NbC-2023
3D-visualization of electronic charge density deformations
in Cu2O-cuprite
ABSTRACT
Present
in different high temperature superconductors, the cuprite Cu2O is a compound
particularly interesting in terms of charge density.
The
analysis was done on the basis of the XRD data of R. Restori et
al. (1986 (1)). To apply the Direct Multipole
Analysis, a reference model compatible with the experimental data is required.
This reference model must follow physical criteria (K. Kurki-Suonio (1977 (9)). Once this reference model is determined, the
analysis can be done.
The first striking feature
in this compound is the remaining metallic character of Cu+ in the
Cu2O lattice. A part of the charge density of Cu+ is free and can
either be used to elaborate the O2─ lattice or be used to fill up
the different empty spaces.
The second striking feature
is the construction of the lattice of O2─ ions alone with a
stabilizing potential due to the environment. The bonding between the O2─
ions is established by the ev2 empty spaces having the same site symmetry than
Cu+.
Finally,
Cu2O is a peculiar compound in which metallic behaviour of Cu+ and
high covalently bridges including the empty spaces necessary to build the O2─
ion lattice, are combined to create the Cu2O lattice.
Introduction
L’étude de la structure de la matière
ne se résume pas aux positions atomiques seules. Il est important de connaître
l’ensemble de la répartition de la densité électronique dans tout le réseau
cristallin. En particulier, les accumulations de charge en dehors des positions
atomiques sont tout aussi importantes que les alentours des sites atomiques.
Alors, la recherche de ces accumulations en dehors des pics atomiques contribue
également aux propriétés physiques ou chimiques des différents composés.
C’est dans cette optique que nous
avons entrepris l’analyse de Cu2O.
La cuprite Cu2O a fait l’objet de multiples
études. Comprendre sa répartition de charge à l’intérieur de son réseau est une
étape incontournable vers la compréhension des supraconducteurs à haute
température.
L’analyse des données RX a été réalisée
jusqu’alors sur la base d’affinements à l’aide de modèles théoriques
multipolaires et entre autres par R. Restori et al. (1986 (1)),
J.M. Zuo et al. (1999) (2)), S.G. Wang et al. (2000 (3)),
T. Lippmann et al. (2000 (4)), P. Coppens et al. (2005 (5)), …. Ces modèles se sont avérés non concluants
d’après les auteurs eux-mêmes.
D’après les résultats reportés dans
la littérature, la nature des liaisons entre les différents atomes reste
confuse.
En appliquant la méthode d’Analyse
Directe Multipolaire (K.Kurki-Suonio
(1968 (6))), nous allons exposer une nouvelle
façon d’aborder le problème.
Nous avons utilisé les données RX de Restori et al. (1986
(1)) pour conduire une Analyse Directe
Multipolaire et sa visualisation. Cette analyse des données ne requiert pas de
modèle sophistiqué, seule une référence « correcte » qui doit suivre
des critères physiques (K. Kurki-Suonio
(1977 (9)) permet de mettre en valeur la région
de valence atome par atome.
Le but de cette analyse est de
déterminer les déplacements expérimentaux des charges à l’intérieur du cristal
et de les visualiser à l’aide de programmes adaptés à la détection individuelle
des contributions atomiques expérimentales et pour tout autre point
d’intérêt.
Déficience du formalisme
conventionnel des facteurs de structure pour Cu2O (7), (8)


Caractéristiques de la structure de Cu2O
La cuprite Cu2O cristallise dans le
groupe spatial cubique P n![]() m (n° 224 T.I.).
m (n° 224 T.I.).
Il y a 2 Cu2O par maille cubique.
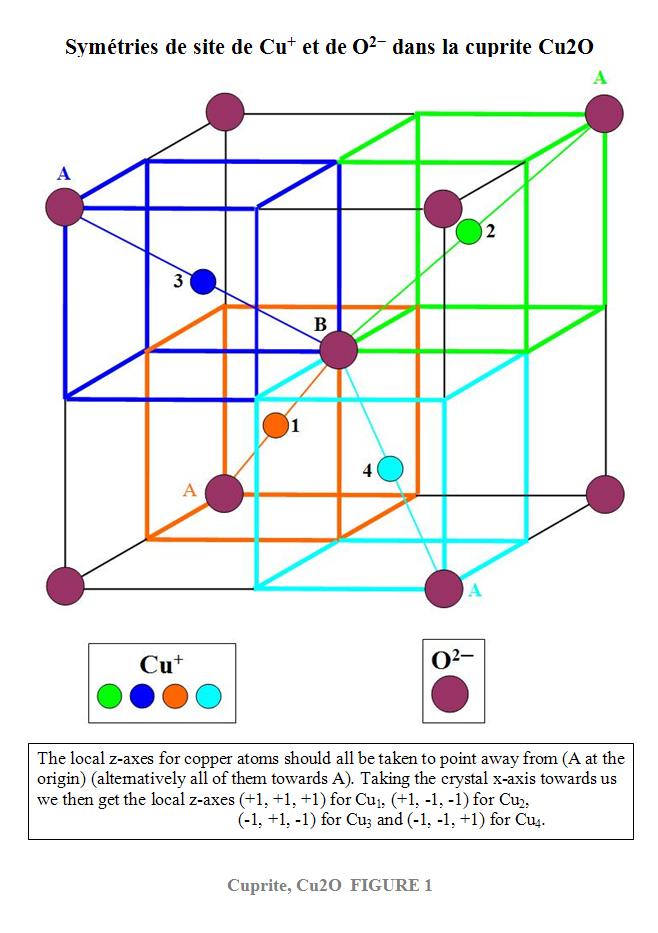
Les 2 oxygènes (figure 1) sont disposés en cube centré et 4
Cu disposés tétraédriquement au centre des cubes partiels de côté a/2 (de
couleur associée aux cubes correspondants d’arête a/2) formant entre eux un
réseau de cube à faces centrées.


Dans la représentation des atomes de Cu aux points d’un
réseau cubique à faces centrées (figure 2),
nous avons pour les positions atomiques (T.I.),
Oxygène : ![]() 3m (2a) 1/4 1/4 1/4 ; 3/4 3/4 3/4.
3m (2a) 1/4 1/4 1/4 ; 3/4 3/4 3/4.
Cuivre : ![]() m (4b) 0 0 0 ; 0 1/2 1/2 ; 1/2 0 1/2 ; 1/2 1/2 0.
m (4b) 0 0 0 ; 0 1/2 1/2 ; 1/2 0 1/2 ; 1/2 1/2 0.
Et 2 types d’espaces vides
Espace vide « ev2 » : ![]() m (4c) 1/2 1/2 1/2 ; 1/2 0 0 ; 0 1/2
0 ; 0 0 1/2.
m (4c) 1/2 1/2 1/2 ; 1/2 0 0 ; 0 1/2
0 ; 0 0 1/2.
Espace vide « ev4 » : ![]() 2m (6d) 1/4 1/4 3/4 ; 3/4 1/4 3/4 ; 1/4 3/4 3/4 ;
2m (6d) 1/4 1/4 3/4 ; 3/4 1/4 3/4 ; 1/4 3/4 3/4 ;
3/4 3/4 1/4 ; 3/4 1/4 1/4 ; 1/4 3/4
1/4.
La figure 3 concerne le cube d’arête
a/2 de centre ev2 (1/2 1/2 1/2)
possédant 6 sommets d’espaces vides sites ev4 et 2 sommets oxygènes. Par
les oxygènes passe le plan (1-10) et contient 2 sites ev4. Les autres sites ev4 bleus sont dans le plan (110).

La figure 4 place le Cu1(000)
au centre du cube. Les oxygènes sont à ±(1/4, 1/4, 1/4 ). Cette
représentation est utilisée pour la visualisation 2D et 3D.
Pour la
visualisation,
nous plaçons le point d’intérêt étudié au centre de la vue 3D par exemple Cu1 ou OB .Cela va nous aider à interpréter les différentes
visualisations ainsi qu’à reconstruire à partir des plans de coupe de chaque
atome (représentation multipolaire) l’ensemble d’un plan du réseau
cristallin.
Sur certaines visualisations, nous avons indiqué quelques
positions de sites.
I-Analyse Directe Multipolaire
L’Analyse Directe Multipolaire
procède depuis la représentation des phénomènes jusqu’à leur interprétation et
leur compréhension.
Par contre, l’emploi des modèles
multipolaires théoriques signifie toujours de calquer un modèle théorique comme
un tout et non de rechercher des paramètres individuels.
Le modèle théorique
conduit ensuite à procéder à l’ajustement
des paramètres. Un ajustement
est jugé bon s’il permet de confirmer l'interprétation théorique originale et
d’attribuer des «valeurs expérimentales» à ces paramètres. (voir SYMPOSIUM
FRANCO-FINLANDAIS SYMPOSIUM).
En général, après des affinements par
least squares, modèle théorique et paramètres thermiques ne sont pas ancrés sur les données expérimentales.
1) Modèle de référence correct
Il faut que les positions atomiques
et les paramètres thermiques d’un atome ainsi que les paramètres de 2 atomes
différents soient indépendants. Pour assurer l’indépendance des paramètres, il
faut utiliser la notion d’orthogonalité.
Cette orthogonalité est atteinte par
l’orthogonalité des composantes Ynmp (θ, φ) d’un
développement multipolaire associé à un
système local spécifique direct d’axes, système adapté à la symétrie de
site du point analysé.
La méthode d’analyse utilisée permet
de séparer ainsi les différents paramètres physiques au moyen des différentes
composantes multipolaires expérimentales (K. Kurki-Suonio (1977 (9)).
Excepté pour les composés du genre
LiH ou LiD (Vidal et al. (1992) (14)) LiH-et-LiD, les paramètres thermiques des atomes
doivent être adaptés aux mouvements des cœurs atomiques de façon à donner les
estimations les meilleures des termes résiduels. Ceci est atteint par une
procédure de calcul qui ajuste leurs valeurs itérativement.
Pour respecter leurs sens physiques,
la représentation des paramètres thermiques doit suivre impérativement la
représentation 4 TiO2-rutile (K. Kurki-Suonio et al. 1980 (10))
soit :
Représentation 4 à partir des déformations
en système local d’axes pour laquelle
D = [Bave + ΔBpr(1/2(3cos2θ─1))
+ ΔBna(sin2θcos2φ)](sinθ/λ)2 où θ et φ sont les
coordonnées sphériques des vecteurs du réseau réciproque dans les axes
thermiques principaux.
Bave est relié à
l’harmonique sphérique d’ordre zéro,
ΔBpr (1/2(3cos2θ─1)
à l’harmonique (2 0 +), où ΔBpr est la « prolateness »,
et ΔBna (sin2θcos2φ)
à l’harmonique (2 2 +), où ΔBna est la non axialité.
Ces correspondances ne sont valables
que si les coordonnées atomiques sont prises dans le système de coordonnées
spécifiques adapté à la symétrie de site locale formée par les axes principaux
de l’ellipsoïde thermique.
Pour Cu : z local
est suivant la direction [111]
y
local est suivant la direction [1-10]
et x local est suivant la
direction [11-2].
Dans un cristal cubique, une direction [uvw] est
perpendiculaire à un plan (uvw) de mêmes indices.
Pour l’oxygène, les axes locaux xyz sont les axes xyz du
cristal.
Dans le cas de Cu2O :
-
pour
O, le site de symétrie ![]() 3m indique une agitation thermique isotrope
3m indique une agitation thermique isotrope
-
pour
Cu, le site de symétrie ![]() m indique une agitation thermique anisotrope
m indique une agitation thermique anisotrope
Ainsi, pour O et Cu, les
critères physiques imposent :
-
Pour les 2 ions, la composante d’ordre 0 doit être nulle à
l’origine (pour assurer la coïncidence des axes locaux avec les axes de
symétrie de l’atome) (K.
Kurki-Suonio (1968 (6)), M. Kara et al. (1981(11)),
-
Pour Cu, en plus, la composante d’ordre 2 (20+) (dérivée seconde)
doit être aussi plate que possible pour les premières valeurs près de r = 0.
Avec ces critères physiques, nous
commençons l’affinement par itération afin d’ancrer
le modèle de référence correct sur les grandeurs expérimentales.

La figure 5 montre les résultats
obtenus avant affinement itératif. Elle
montre la non-conformité des δρnmp(r) avec les critères ci-dessus de K.
Kurki-Suonio (1977 (9)). Comme les positions atomiques
sont imposées par la géométrie du cristal, ce sont les paramètres thermiques
des 2 ions Cu+ et O2─ qui sont à affiner sous forme Bave,
et en plus pour Cu+ ΔBpr
(20+) .
En quelques cycles d’itération, le
modèle théorique de départ est devenu le modèle de référence correct en
adéquation avec le cristal.
Nous sommes assurés que le modèle de
référence correct déterminé est compatible avec les données expérimentales.
Le modèle de référence correct
utilisé correspond à :
pour Cu+ : Bave =0,45945 Å2
ΔBpr = ─0,15949 Å2 ΔBna = 0 Å2
pour O2─ : Bave = Biso= 0,37179 Å2.
Le Cu+ vibre plus que
l’oxygène car nous verrons par la suite qu’une partie de sa distribution de
charge s’est désolidarisée du noyau atomique (électrons libres). La spécificité
de l’ion O2─ s’affirme en
créant de fortes liaisons entre oxygènes
ce qui restreint son mouvement. Dans Cu2O, les électrons de conduction de Cu+
sont canalisés par la présence du réseau des oxygènes. L’oxygène fait son
propre réseau, comme nous l’avons déjà observé (Vidal et al. BeO, MnO,
CoO , NiO, TiO2, SiO2, ….). Réalité
expérimentale de BeO - CERIMES - Vidéo - Canal-U Figure 1 Figure2 Figure 3 TiO2-rutile SiO2-stishovite
2) Facteurs de diffusion atomique des
ions du cristal
L’analyse consiste essentiellement à
l’interprétation des différences entre les données Fobs et les
facteurs de structure théoriques du modèle de référence correct Fref
en terme de développement multipolaire.
Le modèle de référence correct doit
suivre des conditions mathématiques et des critères physiques pour être
compatible avec les grandeurs observées (K. Kurki-Suonio (1977 (6)).
Les conclusions sur les transferts de
charge sont basées essentiellement sur les hypothèses concernant la validité
des facteurs de diffusion théoriques. La contribution de la densité
électronique d’un atome se retrouvera dans son facteur de diffusion atomique.
Pour O2─, l’ion à l’état
libre n’est pas stable, il requiert un potentiel extérieur pour être
stabilisé. En général, c’est le rôle des cations environnants qui permet la
stabilisation de O2─. Les facteurs de diffusion de Watson (+2well)
(1958 (12)) et Sanger (1969 (13)) ont donné des résultats corrects dès lors que
l’ion O2─ était concerné.
Garder le même facteur de diffusion atomique
f(b) pour l’oxygène permet de comparer le
comportement de l’oxygène dans divers composés.
Pour Cu+, le facteur de
diffusion provient des Tables Internationales (IUCr).
Nous utilisons les facteurs de
diffusion atomique sous forme d’une combinaison linéaire de gaussiennes. Pour respecter la signification physique de f (b),
![]()
Il
faut avoir f (b) = 0 pour b→∞. Il faut exclure toute solution avec des B (I)
négatifs ou avec f (b) développé avec une constante.
L’« Analyse Directe
Multipolaire » est basée sur l’utilisation d’un modèle de référence
asymptotiquement correct dans l’espace réciproque.
Dans les calculs d’obtention de la
référence correcte, une représentation gaussienne asymptotiquement ajustée des facteurs
théoriques de diffusion atomique est utilisée pour tenir compte du terme
résiduel.
Pour Cu2O, le cut off est de b=2sinθ/λ =1,623 Å─1. Les
réflexions faibles (221), (322), (421) sont présentes, les réflexions (432) et
(522) n’ont pu être mesurées.
II- Analyse des composantes
multipolaires
1) Etude de la composante d’ordre 0
de la densité de charge atomique

Après obtention de la référence
correcte, cette étude se fait sur la base des courbes 4πr2ρ0(r)
et du nombre Z(r) d’électrons contenus sous le pic atomique (intégrale de 4πr2ρ0(r)).
La
figure 6 présente les résultats obtenus. Le minimum de la courbe expérimentale
4πr2ρ0(r)
donne le rayon de meilleure séparation RM. La hauteur de ce minimum
indique le degré de séparation entre les ions. Le nombre d’électrons compris
sous la courbe 4πr2ρ0(r)
pour r = RM permet de déterminer l’état d’ionisation.
En plus, il est possible de déterminer la nature de la liaison entre ses
voisins.
En partant du principe que les
déformations électroniques sont des quantités relativement petites par rapport
au terme sphérique, le nombre d’électrons à l’ordre zéro donne une première
idée sur la nature du composé à étudier.
Pour Cu+, la courbe
expérimentale montre une légère contraction par rapport à la courbe théorique.
Pour O2─, la hauteur du minimum révèle une séparation faible des
autres atomes bien que le minimum expérimental soit plus bas que le théorique.
En général, la hauteur de ce minimum
indique la nature de la liaison. Pour un minimum très prononcé, la liaison a un
caractère ionique marqué. Pour une hauteur importante, le caractère covalent ne
fait pas de doute.
Il existe un troisième cas qui
correspond à un comportement métallique. Après le rayon de meilleure séparation,
la courbe expérimentale 4πr2ρ0(r) adopte un tracé
parabolique. Tel est le cas pour Cu+ . Ceci indique que la densité de charge n’est
plus attachée à l’atome et prend alors un comportement d’électrons libres comme
dans les métaux (voir figure 1 du SYMPOSIUM
FRANCO-FINLANDAIS SYMPOSIUM).

Sur la figure 7, Cu+ a
son minimum pour RM = 0.95 Å, au-delà de ce rayon, la densité
radiale remonte doucement suivant une parabole. Cu+ a donc gardé
un caractère métallique à l’intérieur du réseau de Cu2O. Pour RM = 0.95 Å, le nombre
d’électrons correspondant à la sphère de rayon RM est de 26,36 e─.
Pour O2─, son rayon de
meilleure séparation est de RM = 1.10 Å et le nombre d’électrons
correspondant à la sphère de rayon RM est de 9,06 e─ ,
valeur récurrente pour O2─, cœur de 9 e─ .
Sur la base des figures 6 et 7, les
rayons choisis pour les sphères
partitives de Cu+ et O2─ nécessaires aux calculs des δfnmp
(b) ont été RCu = 1,30Å et RO = 1,30Å. Ces valeurs
sont généralement plus grandes que les rayons de meilleure séparation définis
par le minimum de ces courbes afin d’étudier les recouvrements de densité
électronique. Ce rayon « trop grand » s’étend clairement dans la
région des premiers voisins mais contient le modèle de déformation plus
complètement.
2) Etude de l’ensemble des
composantes multipolaires dans l’espace réciproque
Les composantes δfnmp (b)
représentent les déviations par rapport au modèle de référence correct des
facteurs radiaux de diffusion atomique. La visibilité de ces déformations est
optimale dans l’espace réciproque. Dans cet espace, les erreurs se concentrent
autour du point de cut off. Tout fait observé entre b = 0 et le cut off est
significatif et d’origine électronique.
Nous avons calculé les composantes
multipolaires δfnmp (b) jusqu’à l’ordre 8.
Pour Cu+ : les seules
composantes multipolaires compatibles avec sa symétrie de site sont :
(2λ, 3μ,
+) : (00+), (20+), (40+),
(43+), (60+), (63+),
(66+), (80+), (83+),
(86+)
Pour O2─ : les seules
composantes multipolaires compatibles avec sa symétrie de site sont :
(harmoniques
cubiques) : (000), (300), (400),
(610), (700), (800)


La figure 8
concerne Cu+ , seuls sont donnés les tracés jusqu’à l’ordre 6.
La figure 9
concerne O2─, seuls
sont donnés les tracés jusqu’à l’ordre 7.
Sur la base des courbes des
composantes δfnmp (b), nous pouvons déterminer les déformations les
plus intenses responsables des propriétés du composé.
Une fois les composantes
multipolaires significatives déterminées, nous pouvons passer dans l’espace
direct.
3) Etude de l’ensemble des
composantes multipolaires dans l’espace direct
Les courbes δρnmp(r) donnent les directions des déplacements
de charge dans le réseau. En même temps, nous pouvons calculer la charge sous
chaque lobe positif des harmoniques sphériques ce qui nous donne la force des
déplacements.
Pour visualiser et rendre
l’interprétation plus facile, les mêmes composantes que pour δfnmp(b) sont aussi représentées dans l’espace direct.
Nous avons calculé les composantes δρnmp(r) pour les 2 ions jusqu’à l’ordre
8.


La figure 10
concerne Cu+, seuls sont donnés les tracés jusqu’à
l’ordre 6.
La figure 11
concerne O2─, seuls
sont donnés les tracés jusqu’à l’ordre 7.
Les figures 10 et 11 montrent que les
critères sur les paramètres thermiques sont respectés.
Les composantes multipolaires δrnmp(r)
indiquent
les accumulations de charge autour des sites atomiques par rapport au modèle de
référence correct.
Pour Cu+, au-delà de RM
(Cu+), la mobilité de la densité électronique est plus conséquente
(figure 10).
Pour O2─, la figure 11
indique que les déplacements sont importants au-delà de RM (O2─).
La normalisation utilisée est telle
que les surfaces sous les courbes donnent le nombre d’électrons sous les lobes
positifs des harmoniques sphériques pour des rayons Ri choisis. Ces
nombres d’électrons ”multipolaires” servent comme caractéristiques intégrales multipolaires des distributions
de charge ionique (voir tableau ci-dessous où r0 est le rayon du
premier lobe significatif).
Tableau 1

Pour Cu+ , les
composantes les plus significatives sont
(00+), (20+), (43+), (60+), (66+).
Pour O2─, ce sont les
composantes (000), (300), (610).
III- Analyse multipolaire des
espaces vides ev2 et ev4
Pour l’Analyse Directe Multipolaire, le système local direct
est adapté à la symétrie du site.
Pour l’étude des espaces vides :
espace vide ev2 ( ![]() m ) comme pour le cuivre
m ) comme pour le cuivre
espace ev4 (![]() 2m ) : z local
est suivant la direction z [001],
2m ) : z local
est suivant la direction z [001],
y local
est suivant la direction [110] et x local suivant la direction [1-10].
Pour ev2, la symétrie de site est identique à celle de Cu+,
par conséquent, les harmoniques sphériques suivront :
(2λ, 3μ, +) : soit 00+ , 20+, 40+, 43+ , 60+ , 63+ , 66+ ,
80+ , 83+ , 86+ jusqu’à l’ordre 8.
Pour ev4, la symétrie de site donne
pour harmoniques sphériques : (2λ, 4μ+1, +) et (2λ+1, 4μ+2, +) :
soit 00+ , 20+ , 32+ , 40+ , 44+ ,
52+ , 60+ , 64+ , 72+, 76+ , 80+ , 84+ , 88+ .
Nous avons calculé pour ces 2 espaces
« vides » les composantes multipolaires des facteurs de diffusion fnmp(b)
et les composantes multipolaires des densités radiales de charge ρnmp(r) jusqu’à l’ordre 8.
Pour les fnmp (b), une
sphère de rayon 1,30Å a été choisie comme pour
Cu+ et O2─.
Le nombre d’électrons contenus dans
cette sphère pour l’ordre zéro est le plus significatif de l’ensemble des composantes.
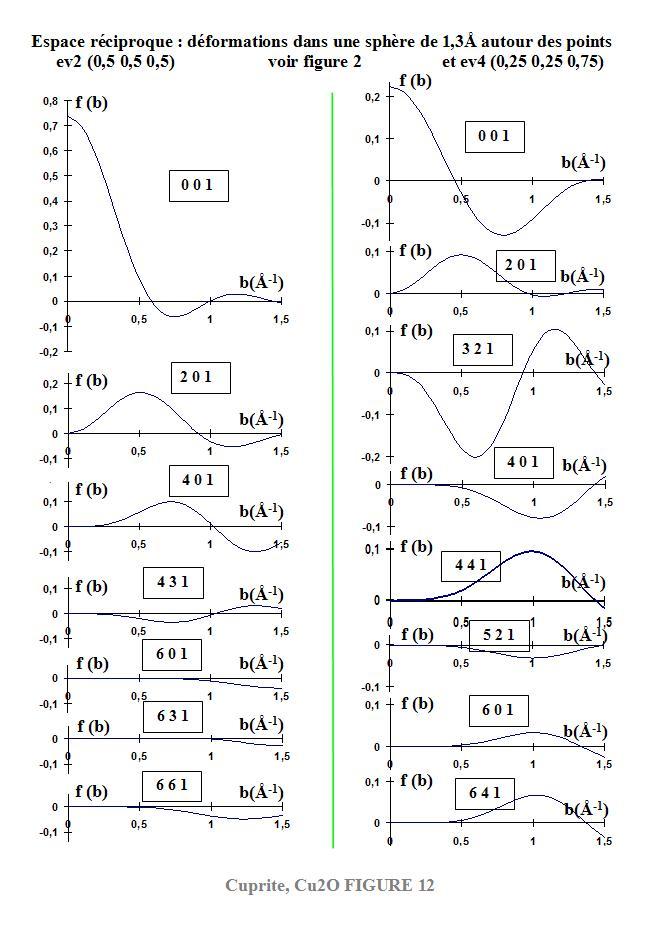
Sur la figure 12, nous avons reporté
les composantes fnmp (b) de ev2 et ev4(b) tracées jusqu’à l’ordre 6.
Pour ev2, les composantes 00+, 20+,
40+ sont les plus importantes. Elles indiquent que la direction (111) du
cristal ou z local joue un rôle primordial.
Pour ev4(b), les composantes 00+,
32+, 40+, 44+ sont les plus importantes avec une prépondérance pour la composante 32+.


Les figures 13 et 14 sont relatives
aux composantes ρnmp(r)
tracées jusqu’à l’ordre 6. La force de la composante (00+) de ev2 est de
0,73591 e─ et celle de ev4 de 0,22427 e─.
Pour un groupement ev2-ev4, le
montant de la charge électronique déplacée est d’environ 1 e─.
IV- Visualisation : 2D et 3D de l’analyse directe multipolaire
Ces visualisations se font à l’aide de
planches reprenant les résultats trouvés précédemment. Les plans visualisés
sont des coupes obtenues à partir des surfaces d’isodensités électroniques
calculées autour de chaque point d’intérêt, positions atomiques ou espaces
vides.
Grâce à nos programmes spécifiques
adaptés à la visualisation de la densité électronique, nous pouvons faire des
rotations autour d’axes choisis. Ici, nous avons pris l’axe z du cristal.


La planche 1 concerne la rotation
autour de z du Cu1+. Autour du centre, il y a un déficit
flagrant de densité électronique par rapport à la référence correcte. Nous
travaillons avec (Fobs ─ Fref).
La planche 2 montre différents plans
et le volume de localisation délimité par la surface d’isodensité électronique
0,08 e/Å3. Le demi volume localise les autres isosurfaces.

La planche 3 reprend le processus
précédent pour l’ion O2─. A signaler l’inversion pour la coupe de O2─
entre les plans (1-10) et (110).
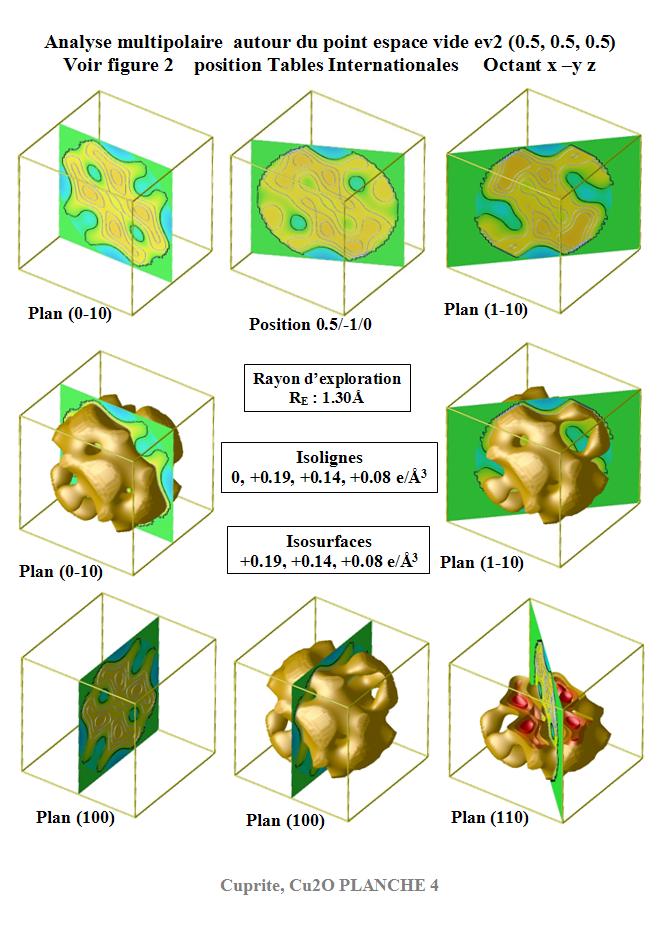
La planche 4 concerne l’espace vide
ev2 (1/2 1/2 1/2) voir fig.2. Dans le plan (1-10), les extensions fines sont
dirigées vers les oxygènes OA et OB, les plus larges vers
les 2 ev4 contenus dans le plan (1-10).


La planche 5 concerne des plans
passant par l’espace vide ev4 (0.25, 0.25, 0.75) appelé b sur la fig.3.
La planche 6 concerne des plans
passant par l’espace vide ev4 (0.75, 0.25, 0.25) voir
fig.3.
Pour ces 2 planches, les différentes
formes suggèrent un volume difficile à reconstruire sur la base des images 2D.


Les planches 7 et 8 montrent les
volumes concernés pour ev4 (0.25, 0.25, 0.75) et ev4
(0.75, 0.25, 0.25).

La planche 9 est relative à la
reconstruction à partir des plans multipolaires (1-10) d’un plan du réseau
cristallin. Le plan cristallin (1-10) passe par les oxygènes ±(1/4, 1/4,
1/4 ) voir fig.4. Ce plan est donné avec ses constituants et montre un
rectangle de hauteur a et de largeur a√2.

La planche 10 concerne l’octant xyz
et montre la genèse des 2 types de ev4. Dans les cubes partiels des 2 types de
ev4, ev4 est au centre du cube correspondant. Les accumulations de charge
importantes font suite aux « pattes » et sont une part de ev2. Nous
voyons l’imbrication entre ev2 et ev4.

La planche 11 visualise les plans
individuels (110) passant par les différents sites et espaces vides. Seules les
coupes de Cu+, ev2 et ev4 (0.75, 0.25, 0.25) sont présentes dans le
plan (110) du réseau cristallin.

La planche 12 montre l’environnement
de OB vu dans 2 octants différents. Les structures
« tripodes » sont différentes selon que l’on regarde vers Cu1
ou ev2. Le cube partiel présente les différentes situations observées.

La planche 13 est relative à la
reconstruction à partir des plans multipolaires (110) d’un plan (110) du réseau
cristallin. Dans ce plan également, l’interpénétration des ev2 et ev4 est très
claire.
V- Visualisation : 2D et 3D de la représentation Fourier
Cette visualisation n’est qu’une
illustration des cartes et volumes multipolaires dans la maille
cristalline.
L’octant x –y z a été choisi car il inclut
tous les atomes (Cu et O) ainsi que les espaces vides.


La planche 14 présente les divers plans (1-10) centrés sur
chacun des atomes ou chaque type d’espace vide.
La visualisation à partir de l’analyse multipolaire permet de
reconnaître chaque constituant du réseau cristallin. Seul ev4 (0.75, 0.25, 0.25)
ne figure pas sur cette planche. Il appartient au plan (110) du cristal.
Sur la planche 15, nous avons changé
d’octant afin de visualiser ev4 (0.75, 0.25, 0.25). Les interactions entre les différents
constituants sont visibles.
Dans le plan (110), la figure
relative à l’oxygène n’est pas dans le plan du réseau cristallin passant par Cu1(000).
Il appartient au plan (1-10) du réseau cristallin.


Sur la planche 16, nous avons représenté les plans
cristallins (1-10) et (110) pour un cube d’arête 2a afin de suivre les
accumulations de charge à travers le cristal. L’oxygène est absent du plan
(110).
Cette planche est à comparer avec les planches 9 et 13 des
reconstructions à partir des éléments de l’analyse multipolaire. Ceci montre à
quel point l’analyse multipolaire retrace l’ensemble du réseau point par point.
Les rectangles en pointillés présents sur la planche 16
délimitent une maille de Cu2O.
La planche 17 est relative à une seule maille de Cu2O, soit
un cube d’arête a.

Pour la planche 18, nous restons dans l’octant x y z avec le
plan (110). En déplaçant ce plan, nous retrouvons les oxygènes OA et
OB absents au niveau du plan passant par Cu1(000).
Les volumes correspondants montrent les rangées de ev2 et la
structure des accumulations de charge
électronique autour de l’oxygène OB.


Sur la planche 19, le déplacement suivant z d’un plan (001)
précise la forme des accumulations de charge
électronique dans une maille.
Pour la planche 20, nous avons repris la planche 19 en
enlevant le plan (001). On peut alors suivre la construction des accumulations
de charge.

Sur la planche 21, revenons à l’octant x –y z centré sur ev4 (0.75, 0.25, 0.25),
les coupes montrent l’imbrication entre ev2 et ev4. Les
extensions en forme de « pattes » appartiennent à ev4 et
prolongent ev2.
VI- Remarques
Cu2O est remarquable pour 2 raisons principales.
a) Dans ce composé, le cuivre garde
un caractère métallique à l’intérieur d’un corps non métallique (fig.7). Cu
métallique appartient à un réseau cubique faces centrées d’arête 3,61Å. Inclus
dans Cu2O, Cu est en position cfc, mais l’arête a pour valeur 4,267 Å soit une
augmentation de la maille cfc de 18,2%.
b) L’oxygène O2─ reste
fidèle à lui-même : il construit son propre réseau d’O2─ en
captant une partie de la densité électronique de Cu+.
1) Le cuivre possède une densité électronique capable de se désolidariser
de l’atome. Cette partie « libre » de sa densité de charge s’infiltre
ou diffuse vers les espaces vides. Majoritairement, cette densité
« libre » va occuper une symétrie de site analogue à celle du Cu+
(![]() m) c’est-à-dire ev2. Les ev4 ne représentent que l’excédent
de densité « libre » qui va jusqu’à s’étendre dans le 2ème
type d’espace libre ev4.
m) c’est-à-dire ev2. Les ev4 ne représentent que l’excédent
de densité « libre » qui va jusqu’à s’étendre dans le 2ème
type d’espace libre ev4.
Cette densité de charge
« libre » de Cu+ est si importante que même après
captation d’une partie par l’oxygène, il reste suffisamment de densité de charge pour combler l’espace vide
ev2 et déborder dans ev4.
2) L’oxygène confirme son comportement déjà étudié dans plusieurs
composés. Il constitue son réseau propre à 10 e─ et pour cela, il va
annexer une partie de la densité de charge de ses voisins et créer un potentiel
dans lequel il est stable.
Pour visualiser cet état de fait, la
représentation à base d’Analyse Directe Multipolaire analyse bien la situation
individuelle de la densité électronique autour de l’oxygène.
Sa déformation souligne les
directions dans lesquelles l’oxygène va puiser de la densité électronique pour
réaliser son réseau spécifique.
En particulier, sur les planches 3 et
12, les excroissances en forme de tripodes indiquent les directions de
captation de la densité électronique de l’oxygène.
Du fait des comportements de Cu+
et d’O2─, les espaces vides ev2 et ev4 prennent un relief
particulier. L’ev2 représente un important transfert de charge vers un espace
vide.
Les ev4 ne représentent que
l’excédent de densité « libre » qui va s’étendre dans le 2ème
type d’espace libre.
Etant donné la nature de ev2, ev4 ne
sera que le prolongement des ev2 vers l’espace libre restant de symétrie de
site différente. Il est donc naturel de trouver que ev2 et ev4 sont liés. L’ev4
n’est qu’un écoulement de ev2 vers un espace vide.
L’imbrication de ev2 et ev4 est
nettement visible sur la planche 21. On conçoit bien l’arrangement des
accumulations de charge autour des sites étudiés.
Les 2 types de ev4 ne sont différents
que par leur position dans l’espace. En terme de quantité de charge déplacée,
ils sont identiques. Seule la répartition suit les symétries de site imposées
par le réseau cristallin.
Cette analyse n’a été possible que
par l’Analyse Directe Multipolaire. En utilisant l’Analyse Directe
Multipolaire, nous avons pu détecter les configurations et les étendues des
divers composants du réseau cristallin.
L’étude des espaces vides nous a
permis de comprendre la constitution du réseau cristallin point par point.
La visualisation des accumulations de
charge donne les directions dans lesquelles ces accumulations vont s’étendre et
donc créer les différents liens entre les atomes.
La reconstitution à partir des
représentations directes multipolaires montre l’avantage de l’Analyse Directe
Multipolaire par rapport à la représentation Fourier classique sur l’ensemble
de la maille.
Le « Fourier » classique
est aveugle et ne donne qu’une vue globale de la maille en ignorant quel
composant apporte sa contribution en un point considéré.
Il est alors difficile sur la base du
seul « Fourier » classique de connaître qui fait quoi dans le réseau
cristallin. A défaut de savoir le rôle de chacun dans le réseau, des modèles de
densité électronique sont alors élaborés. Il est délicat de générer un modèle
fiable en l’absence d’éléments spécifiques à la nature des répartitions
électroniques individuelles.
Au contraire, l’Analyse Directe
Multipolaire permet de déterminer le rôle de chaque composant dans le réseau
cristallin indépendamment de tout modèle sophistiqué.
Les propriétés de Cu2O sont liées aux
caractères de Cu+ resté métallique et aux oxygènes qui cherchent à
constituer un réseau spécifique d’O2─ seuls. La planche 20 montre
l’arrangement en feuillets parallèles à une face (111) qui constitue une
caractéristique de Cu2O.
La coexistence de ces 2 composants
crée un composé qui sert de base à l’élaboration de supraconducteurs à haute
température.
Références
(1) R. Restori &
D. Schwarzenbach, Acta Cryst.
(1986) B42 201-208.
(2) J.M. Zuo, M. Kim,
M. O’Keeffe & J.c.H. Spence, Nature (1999) vol 401, p.49-52
(3) S.G. Wang &
W.H.E. Schwarz, Angew. Chem.Int.Ed. (2000) 1757-1762
(4) T. Lippmann & J.R. Schneider, J. Appl. Cryst. (2000)
33 156-167
(5) P. Coppens, B. Iversen, Finn Krebs Larsen, Coordination
Chemistry Reviews (2005) 249 179-195
(6) K. Kurki-Suonio (1968) Acta Cryst.
, A24, 379-390
(7) K. Kurki-Suonio Report series in Physics,
(8) K.Kurki-Suonio
Acta Cryst. (1970) A26,
458
(9) K. Kurki-Suonio (1977) Isr. J.
chem. 16, 115-129; 132-136
(10)
H. & K.
Kurki-Suonio, S. Rosten and R. Sälke (1980) Report series in physics,
HU-P-191,
(11) M. KARA & K. KURKI-SUONIO (1981) Acta Cryst. A37, 201-210
(12) R.E. WATSON (1958)
Phys. Rev.
111, 1108-1110
(13) P.L.SANGER, (1969) Acta Cryst.
A25, 694-702
(14) G. Vidal-Valat, J-P. Vidal, K.
KURKI-SUONIO, R. Kurki-Suonio (1992)
Acta Cryst. A48 46-60 “Evidence
on the Breakdown of Born-Oppenheimer Approximation in the Charge Density of
Crystalline 7LiH/D”
mag3D Figure1 Figure2 Figure3 PRESENTATION SYMPOSIUM FRANCO FINLANDAIS SYMPOSIUM Problème des phases The phase problem DVD Semi-conducteur organométallique organosilicié TiO2-rutile SiO2-stishovite Cu2O-cuprite K2PtCl6 Be-metal bases and 3D views Bases et vues 3D Si-et-Ge CaF2-fluorite NbC-2023