mag3D Figure1 Figure2 Figure3 PRESENTATION SYMPOSIUM FRANCO FINLANDAIS SYMPOSIUM Problème des phases The phase problem DVD Semi-conducteur organométallique organosilicié TiO2-rutile SiO2-stishovite Cu2O-cuprite K2PtCl6 Be-metal bases and 3D views Bases et vues 3D Si-et-Ge CaF2-fluorite NbC-2023
VISUALISATION 3D DES
DISTRIBUTIONS DE CHARGE
DANS LE SILICIUM ET LE
GERMANIUM
Electron
density distribution and 3D visualization
in
silicon and germanium
Abstract
A
study of the charge distribution of Silicium and Germanium was undertaken on the
basis of the RX powder data of Saravanan et al. (2008 (3)).
The
aim of this work is to apply the Direct Multipole Analysis PRESENTATION,
SYMPOSIUM FRANCO FINLANDAIS, SYMPOSIUM and its visualization to evidence the charge
transfers in the valence region of these two compounds.
The
comparison between Si and Ge led to appreciate the differences in the behaviour
of Si and Ge.
The
germanium presents a more complex charge distribution difference than the
silicium. For Ge, the charge distribution extends largely up to the empty
space.
This
feature can explain the different properties of the two compounds.
Introduction
Les motifs de diffraction des RX
donnés par les cristaux peuvent être employés pour trouver l’arrangement exact
des atomes et la répartition des charges à l’intérieur du composé et entre les
atomes.
Le silicium et le germanium possèdent
la structure de type « diamant ».
La maille de Si ou de Ge contient 8
atomes, 4 atomes forment un réseau cubique faces centrées (sites A), les 4
autres occupent en tétraèdre les centres de 4 des 8 cubes partiels de coté a/2
(sites B) dans la maille cubique de coté a.
Coordonnées : 000, ½ ½ 0, 0 ½ ½
, ½ 0 ½ ¼ ¼ ¼ , ¾ ¾ ¼ , ¼ ¾ ¾ , ¾ ¼
¾ (figure 1) .
Si et Ge sont des composés très
étudiés à cause de leurs propriétés semi-conductrices.
S. Cummings & M. Hart (1988 (6)) ont étudié pour Si 2 ensembles de 2 types de
données basées sur les positions des franges de Pendellösung. Leurs conclusions
concernaient surtout la comparaison entre la théorie dynamique de diffraction
et l’expérience.
Z. W. Lu et al. (1993(7)) ont effectué une étude sur C, Si et Ge. Ils ont
essentiellement discuté sur les modèles multipolaires utilisés.
R. Saravanan et al. (2008 (3)) ont entrepris des analyses de Si et Ge sur de
nouvelles données obtenues à partir de poudres. Ces analyses ont porté
essentiellement sur :
- l’affinement multipolaire à l’aide
de modèles multipolaires,
- le maximum d’entropie
Les conclusions de leurs études
concernaient essentiellement une comparaison des différentes méthodes.
A partir des données de R. Saravanan
et al. (2008 (3)), nous avons entrepris une
étude et une visualisation 3D des répartitions de charge différence par l’Analyse
Directe Multipolaire qui évite d’utiliser des modèles a priori.
L’étude 1D permet l’obtention d’une
référence correcte adaptée aux données expérimentales. L’Analyse montre un
comportement de semi-conducteur dans les 2 cas. Par contre, le sens des
transferts de charge ne sont pas identiques pour Si et Ge.
L’étude 2D permet d’utiliser 2
représentations : atome par atome (Analyse Directe Multipolaire) ou 1 ou
plusieurs mailles (Fourier conventionnel).
Outre la présence d’artefacts dans la
distribution de charge différence de Si, l’absence d’accumulation de charge
dans les plans {110} de Si accentue la différence de comportement entre Si et
Ge.
Cette étude nous permet aussi de voir
l’interaction (ou pas) entre les 2 types de sites A et B.
L’étude 3D restitue en volume les
répartitions de charge différence. La comparaison entre Fourier conventionnel
et Analyse Directe Multipolaire permet de déterminer le rôle de chaque atome
dans le réseau.
La différence de comportement entre
Si et Ge est évidente. Le réseau d’accumulation de charge est beaucoup plus
compact et continu dans le cas de Ge, mais pas entre premiers voisins A et B
qui se correspondent par un centre d’inversion.
Les reconstitutions des mailles au
moyen des figures de l’Analyse Directe Multipolaire favorisent la compréhension
des divers réseaux de répartition de charge.
Avant-propos


Comme les structures géométriques de Si
et Ge (positions atomiques) sont analogues à celles du diamant, les remarques
indiquées dans le texte de K. Kurki-Suonio restent valables.
En particulier, le choix de l’origine
au centre d’inversion entre A et B montre que les réflexions de type 222 ne sont
pas interdites et font partie des réflexions inhérentes à l’association des
centres d’inversion avec le groupe spatial.
Pour visualiser le référentiel
utilisé pour analyser les mesures RX, nous avons tracé 3 figures différentes
des mailles cubiques de Si et Ge.

Cette figure valable aussi bien pour
Si que pour Ge a été tracée en plaçant un atome de site A à l’origine. Les
atomes de site B sont placés suivant un tétraèdre à l’intérieur du cube
représentant la maille cubique. Elle montre la représentation la plus
conventionnelle des structures de type diamant, Si , Ge,…
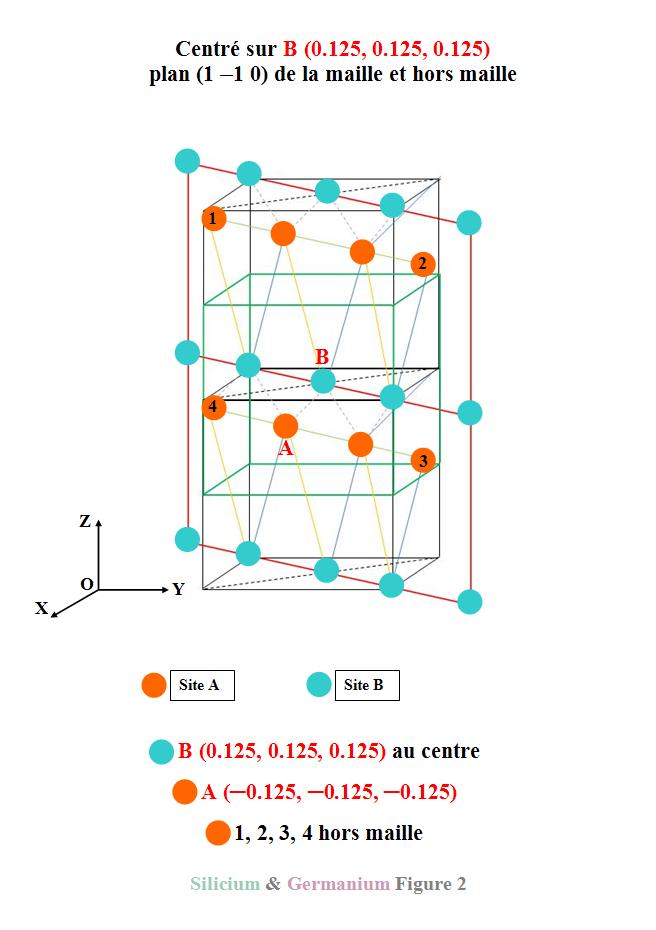
Centrée sur un atome de site B, elle
visualise le plan (1 ─1 0). Les traits
jaunes et bleus indiquent les directions entre les atomes de site A et B
correspondant aux trajets les plus longs. Les pointillés violets tracent les
directions des plus proches voisins entre A et B.
Nous verrons par la suite les
correspondances en terme de répartitions de charge. Le cube en traits verts
représente une maille cubique de Si ou Ge.
Il a fallu considérer 2 mailles
cubiques pour rendre compte de l’aspect du plan (1-10). Ce plan (1-10) joue un
rôle particulier dans la visualisation de la densité de charge différence car
il contient les 2 types de sites A et B.
Il va nous permettre de juger de
l’interaction entre ces 2 sites.

Elle précise la maille que nous avons
considérée en prenant l’origine au milieu des sites A et B les plus proches.
Cette figure fait apparaître la position des 2 tétraèdres des sites A et B
disposés tête bêche.
Entre les sites A et B, il existe un
centre d’inversion. Un exemple de centre d’inversion est le point χ centre du
cube de la figure 3.
C’est sur la base de cette figure que
nous avons mené l’Analyse Directe Multipolaire et le Fourier conventionnel.
Nous y avons associé le tableau de données de base correspondant aux valeurs
des paramètres utilisés en cours d’Analyse.

Dans ce tableau, nous avons
répertorié l’ensemble des données que nous avons utilisées en cours d’analyse.
Nous avons utilisé les données de Saravanan et al. (2008 (3)) obtenues sur des poudres et en plus la valeur du
facteur de structure de la réflexion 222 adaptée aux données de Saravanan et
al. (2008 (3)). Par exemple, la littérature
présente FSi : 222 et FGe : 222 de R.W. Alkire
et al. (1982(4)), et de A. Kazimirov et al.
(2011(5)).
Cristal et Diffraction
Chaque facteur de
structure F(hkl) représente à la fois l’amplitude et la phase du faisceau
diffracté, cette dernière se référant à l’origine choisie dans le cristal.
Dans le cas d’une structure type
diamant, il est intéressant de prendre l’origine sur un centre d’inversion car
les phases des F(hkl) sont 0 ou π (fig.3). Dans la représentation de
la fig.1, les F(hkl) ont pour phases 0 ou ± π/4.
Les facteurs de structure F(hkl) sont
les coefficients de Fourier de la fonction périodique ρ(r)
densité électronique exprimée en nombre d’électrons (quantité d’électricité 1,6
10─19
Coulomb) par unité de volume e─/Å3 (PRESENTATION).
La correspondance entre
F(hkl) et ρ(r) suggère la possibilité d’une méthode Directe d’Analyse
du cristal. C’est cette
voie que nous avons choisie par l’Analyse Directe Multipolaire. Alors, il n’est
pas nécessaire d’essayer d’ajuster des modèles sur les valeurs des hkl
mesurées.
Pour constituer une série de Fourier
tridimensionnelle, les F(hkl) doivent être mesurés pour un domaine continu de valeurs hkl et rangés en fonction de sinθ/λ
croissant. Or le nombre de données mesurées est limité. Il n’y a pas de mesure
au-delà d’une valeur limite en sinθ/λ, d’où l’importance de connaître avec précision
le terme résiduel (bases and 3D views , Bases et vues 3D).
Le terme constant dans la série de
Fourier provient du terme en h=k=l=0 et il est égal à Z0 nombre
total d’électrons dans la maille.
Dans le cas d’une structure type
diamant, il est nécessaire d’inclure la valeur de la réflexion 222 (petit
sinθ/λ mesurable). Les informations sur les déformations atomiques ou ioniques
se trouvent dans le domaine des réflexions en petits sinθ/λ.
Si le terme résiduel est ignoré ou
mal établi (bases and 3D views , Bases et vues 3D), ceci entraîne des erreurs ou des valeurs
biaisées pour les densités calculées.
Au sens de la diffraction
X, les centres des
atomes sont désignés comme les positions des maximums de densité et les atomes
peuvent s’identifier en calculant la concentration de la matière diffusante et
obtenir ainsi le nombre d’électrons associés à chacun d’eux (voir courbes 4πr2ρ0(r)),
ρ0(r) étant le terme sphérique du
développement multipolaire de l’atome considéré).
Comme on travaille en Fexp
─ Fth, avec Fth sphérique par essence (voir littérature
facteur de diffusion de l’atome), les termes non sphériques du développement
multipolaire sont donc directement reliés aux déformations atomiques présentes
et sont des quantités purement expérimentales.
Etude 1D
I- Analyse Directe Multipolaire à 1 dimension
Cette analyse concerne le
développement en composantes multipolaires adaptées à la symétrie du site
considéré. Elle donne, sans modèle, la fonction radiale de la distribution de
charge autour de ce site.
Elle nous permet la détermination de 3
propriétés du composé :
1) la spécification d’une référence
correcte nécessaire pour occulter le pic de diffraction et donc de mettre en
lumière la région de valence,
2) à l’ordre zéro, l’indication du
type de composé (ionique, covalent, métallique,…) par l’allure de la courbe 4πr2ρ0(r)
(voir SYMPOSIUM FRANCO-FINLANDAIS et SYMPOSIUM) et de l’état ionique par le nombre d’électrons sous le pic atomique.
3) Une fois la référence correcte déterminée,
les ordres asphériques du développement multipolaire indiquent le sens des
transferts de charge entre les atomes.
1) Référence correcte

La figure 4 illustre la
non-adéquation des données avec le modèle utilisé pour les séries différence.
Il est manifeste que le terme sphérique du coefficient d’agitation thermique du
modèle ne coïncide pas avec le terme sphérique du coefficient thermique des
données expérimentales. La condition δρ0(r)
= zéro à r =
0 n’est pas respectée (bases and 3D views , Bases et vues 3D),.
En particulier pour Si, la valeur en
r = 0 indique une forte différence entre les 2 coefficients thermiques l’un
venant des données et l’autre venant du modèle. La situation est meilleure pour
Ge mais pas idéale.
2) Ordre zéro du développement multipolaire
Après ajustement des coefficients
d’agitation thermique pour avoir une référence correcte, (bases and 3D views , Bases et vues 3D) nous avons calculé les densités
radiales de charge (ordre zéro) et le nombre d’électrons Z0(r) dans
des sphères concentriques de rayons croissants.


La figure 5 est relative à Si.
L’allure de la courbe 4πr2ρ0(r)
est typique d’un
semi-conducteur (SYMPOSIUM
FRANCO-FINLANDAIS et SYMPOSIUM). Pour mieux visualiser les différences entre le
cristal et la référence correcte, nous avons précisé les courbes dans la région
du rayon de meilleure séparation (minimum de la courbe). L’allure de la courbe
indique que nous ne sommes pas en présence d’un métal car il n’y a pas de
parabole possible partant de l’origine ni pour Si (fig.5), ni pour Ge (fig.6).
Pour Si, RM = 1,609Å RM rayon de meilleure séparation et le nombre Z0
d’électrons dans cette sphère RM est de 13.222 e─
inférieur au 14 e─ de Si.
De façon similaire, la figure 6 donne
les résultats pour Ge qui a aussi un profil de semi-conducteur.
Pour Ge, on a RM = 1,375 Å
pour Z0 (RM) = 30.346 e─ inférieur au 32 e─
du Ge.
Ceci indique qu’une partie de la
distribution de charge se répartit entre les composantes non sphériques et peut
s’étaler en dehors des positions atomiques.
3) Ordres asphériques du développement multipolaire
Notre analyse commence toujours par
une étude dans l’espace réciproque afin de déterminer les composantes
multipolaires significatives. Les calculs sont faits jusqu’à l’ordre 10 mais
nous ne conservons que les composantes réellement présentes.

La figure 7 concerne les composantes
δfnmp(b) des facteurs de diffusion atomique radiaux de Si et Ge.
Dans les 2 cas, nous avons considéré
les composantes jusqu’à l’ordre 6 qui restent significatives.
Les ordres 3 et 4 sont prépondérants
et positifs pour Si alors que l’ordre 3 s’inverse et l’ordre 4 est plus
important pour Ge. Les ordres 6 sont du même ordre de grandeur pour Si et Ge.
On voit déjà que Si et Ge n’ont pas
des comportements identiques.

La figure 8 trace les composantes
multipolaires radiales δρnmp(r) autour de Si et Ge dans l’espace direct ou espace
cristallin.
Ces courbes permettent de vérifier
l’adéquation des coefficients d’agitation thermique entre les données
expérimentales et la référence correcte à l’ordre zéro. La condition δρ0(r)
= zéro à r =
0 est atteinte.
Les composantes asphériques montrent
les transferts de charge dans l’espace direct à l’intérieur du cristal.
Le renversement de la composante
d’ordre 3 indique que les transferts de charge se font en sens inverse pour Si
et Ge alors que les composantes d’ordre 4 et 6 indiquent le même sens de
transfert.
La composante d’ordre K3 négative
pour Si indique que le transfert de charge s’éloigne de la <111>
indiquant qu’il n’y a pas d’interaction entre les sites A et B pour Si.
L’ordre K4 positif indique un
transfert de charge depuis les <111> vers les directions <100>. L’ordre
K6,1 positif souligne un transfert depuis la <110> principalement vers
les directions <111> et plus faiblement vers les directions <100>
voir DVD .

Les nombres d’électrons Z(R)
correspondant aux composantes asphériques sont relatifs aux domaines contenant respectivement 14 e─
pour Si et 32 e─ pour Ge. Les rayons ont été observés sur les
courbes 4πr2ρ0(r) (fig.5 et 6).
r0 est le rayon relatif au
premier lobe indiquant le passage de δρnmp(r) par zéro et RM est le
rayon de meilleure séparation.
Les signes + ou ─ indiquent le sens
des transferts de charge, + du lobe négatif vers
le lobe positif et ─
le sens inverse. Par exemple pour K4,
si la composante 4 est négative, cela implique un transfert de charge depuis <001> au profit de <111>.
Etude 2D
II- Analyse Directe Multipolaire à 2 dimensions : Atome par Atome
L’Analyse Directe Multipolaire à une dimension
est indispensable et incontournable pour avoir une bonne image des déformations
électroniques dans la région de valence. Il n’est pas facile d’associer
mentalement chaque composante multipolaire à l’harmonique sphérique
correspondante. Aussi, nous les présentons dans DVD.
Nous avons donc développé une
représentation en 2 dimensions sous forme de cartes qui sont en fait des coupes
dans la densité électronique différence volumique.
Cette représentation peut s’appliquer
à notre Analyse Directe Multipolaire comme à une représentation de Fourier
conventionnelle sur l’ensemble de la maille.




Les planches 1, 2, 3, 4 retracent la
rotation d’un plan en démarrant par un plan en position (1 ─1 0) jusqu’au plan
(1 1 1).
La planche 1 correspond à une
rotation autour de Z. Les divers aspects de la densité électronique soulignent
la complexité du volume construit autour des sites atomiques Si et Ge.
Les déformations électroniques
semblent inversées entre Si et Ge (voir l’inversion de la composante d’ordre 3
fig.7 et 8).
La planche 2 est la continuité de la
rotation initiée sur la planche 1. Si les plans (100) de Si et Ge semblent
similaires, une petite rotation autour de Z rétablit l’inversion vue
précédemment.
La planche 3 concerne une rotation
horizontale qui nous amène du plan (110) au plan (111). Là encore,
l’inversion des déformations est
nettement visible entre Si et Ge.
La planche 4 est relative au plan
(111). Afin de montrer l’inversion entre les 2 sites A et B, nous avons
présenté le même plan centré sur B puis sur A.
La comparaison entre les plans des
sites A et B met en évidence le centre d’inversion situé au milieu de AB plus
courte distance.

Elle montre différents plans par
volumes tronqués et perpendiculaires à
l’axe Z à différentes hauteurs.
Les différences entre Si et Ge sont
nettement visibles. Les rayons d’exploration ont été choisis pour considérer
les domaines contenant 14 e─ pour Si et 32 e─ pour Ge. Ce
choix permet d’analyser la totalité des répartitions de charge dans Si et Ge.
Pour Si, on observe plutôt des nodules, par contre pour Ge, la densité de
charge est plus étalée.

La planche 6 montre de face
le plan (110). Nous avons utilisé pour cela un changement de référentiel.
Associé au plan (110), nous avons
visualisé en 3 dimensions le volume correspondant à ce changement d’axes. Il
est normal d’avoir plus de déformations, vu le nombre d’électrons, pour Ge (32
e─) que pour Si (14 e─).
Par contre, la reconstitution d’un
volume à partir de coupes n’est pas évidente et peut entraîner des erreurs
d’appréciation.
III- Analyse par Fourier conventionnel à 2 dimensions : maille
Nous avons ensuite procédé à une
illustration des séries différences à l’aide d’une transformée de Fourier
conventionnel. Dans ce cas, toutes les composantes significatives ou non
significatives et le bruit de fond sont inclus dans la représentation Fourier
par essence.

La planche 7 représente les plans
principaux de la structure de Si et Ge.
Là encore, sur le plan (1 ─1 0),
l’inversion entre les déformations de Si et Ge est notable.

Elle représente les plans (110)
d’abord pour une maille puis pour 4 mailles.
L’origine de la maille est prise sur
le centre d’inversion mi-distance entre sites A et B les plus rapprochés. Les
sites A et B et le point χ ont été indiqués. L’accumulation de charge au point
χ n’est relative qu’à Ge. Il est placé au
milieu de la distance entre A et B la plus longue. Egalement, les artefacts
dans Si sont visibles placés en dessous et au-dessus des sites A et B.
En comparant les cartes de Si et Ge,
on voit que l’artefact de Si se trouve dans une zone de très basse densité pour
Ge, ce qui conforte l’interprétation d’artefacts pour Si.
Sur ces plans, on voit qu’il n’y a
pas d’interaction entre les sites A et B les plus proches. Par contre pour Ge,
une forte déformation est présente entre les sites A et B les plus éloignés
(voir les traits jaunes et bleus de la fig. 2).

Cette planche est relative
aux plans (111) autour des sites A et B pour une maille tout en restant centré
sur B. On note le
déplacement des motifs B par rapport aux motifs A. En haut, au centre on voit
bien B (plan B (111)) puis en bas, les 3 A (plan A (111)) dans les directions
de plus courtes distances AB. On en déduit les centres d’inversion entre A et
B.

La planche 10 montre pour le Si, le
plan (110) pour 1 et 4 mailles avec les artefacts. Sur la figure du haut, nous
avons noté les sites A et B ainsi que l’origine de la maille choisie pour que
les phases des facteurs de structure de Si et Ge soient 0 ou π.
- Dans la direction AB passant par
l’origine, il n’y a pas d’accumulation
de charge visible pour Si.
- Le long de AB les plus éloignés, il
n’y a pas d’accumulation de charge. Les accumulations de charge se font plutôt
suivant la direction de l’axe Z et plus généralement suivant les directions
<100> reliant les atomes d’un même
type de site A ou B.

Elle concerne toujours le Si et le
plan (110). Afin de supprimer la persistance rétinienne gênée par les
artefacts, nous avons occulté les artefacts en les recouvrant par des surfaces
bleues.
Les traits violets correspondent aux
traits pointillés violets de la figure 2 alors que les traits rouges
visualisent les traits bleus et jaunes de la figure 2.
Le rectangle noir correspond à une
maille du réseau de Si et nous avons marqué l’emplacement des espaces
« vides » correspondant aux artefacts.

Cette planche est dévolue au Ge. Là aussi,
nous avons représenté une maille et 4 mailles.
Les 2 types de sites A et B sont
notés ainsi que l’origine de la maille et le point χ au milieu de
l’accumulation de charge reliant les sites A et B les plus éloignés.
Entre les sites A et B les plus proches,
une absence d’interaction est notoire.

C’est la carte de Fourier
conventionnel s’étendant sur 4 mailles dans le plan (110) dans Ge.
Les sites A et B et l’origine de la
maille considérée sont signalés. Les directions reliant les A et B les plus
éloignés sont marquées en rouge (traits bleus et jaunes de la figure 2). Le
point χ se trouve au milieu de ces
directions et au milieu de la maille cubique de la figure 3. Les coordonnées de
quelques points χ sont indiquées sur le plan.
Le réseau d’accumulation de charge en
zigzag est une caractéristique du réseau de Ge.
Etude 3D
IV- Analyse par Fourier conventionnel à 3 dimensions : maille
Une importance particulière sur le
choix des valeurs des isosurfaces est mise en œuvre pour représenter au mieux
les interactions.
Dans ce but, nous avons représenté un
certain nombre de volumes.

Dans la représentation Fourier conventionnel, la planche 14
regroupe des vues de Si et Ge.
Dans la planche 14, les vues
correspondant au plan (001) permettent de comparer les densités de charge
différence entre Si et Ge. La structure noduleuse de Si édifie des arrangements
cubiques autour de la position de Si.
Autour de Ge, la densité de charge
autour de la position atomique est plus continue et forme des réseaux
s’étendant dans la maille. Cette planche montre la nette différence entre les
réseaux de Si et de Ge. On retrouve cette différence à chaque différente coupe
du volume.

La planche 15 montre le réseau
spécifique de Ge. La planche 15 est établie dans un
référentiel différent de façon à voir le plan (110) de face.
Dans la planche 15, le volume 3D sur
4 mailles et différents plans de la structure de Ge sont représentés. Le volume
3D montre la genèse de la structure en zigzag et sa représentation 3D.

Dans la planche 16, nous avons isolé
une seule isosurface de Ge pour mieux rendre compte en 3D de l’élaboration du
volume continu à l’intérieur du réseau de Ge.
Nous avons marqué un point χ qui,
étant un centre d’inversion, joue le rôle d’un nœud important dans la structure
volumique de la densité de charge différence de Ge.
En prenant différentes tranches du
volume, on se rend compte de la complexité du réseau de Ge.

C’est une planche de Si équivalente à
la planche 15 pour Ge. Le changement de référentiel nous permet de visualiser
de face le plan (110).
Aucune structure en zigzag n’est
visible. Par contre, les accumulations de charge révèlent une structure
noduleuse suivant les directions <100> (plan (001)).
V- Analyse Directe Multipolaire à 3 dimensions : site par site
Les planches suivantes ont pour but
de montrer qu’à partir de la représentation site par site, il est possible de
reconstruire la maille cristalline.

Pour Si, nous
avons vu que le plan (110) ne présentait pas d’interaction notoire entre les
sites A et B. Par contre, les interactions se situaient entre les atomes de
même site. La planche de reconstruction de Si concerne donc le plan (100) dans
lequel il n’y a que des sites de même type ici B.
En superposant les vues obtenues par
la représentation multipolaire, on s’aperçoit qu’il existe des interactions
entre les atomes des sites B formant des carrés autour des Si et plus
généralement dans l’espace, un arrangement cubique de nodules.
En bas, la figure provient de la
représentation Fourier conventionnel et révèle la position des artefacts
centrés sur des espaces « vides ».

Pour Ge , nous
avons reconstruit le plan (110) à partir des figures de la représentation
multipolaire.
En plus des volumes centrés sur les sites
A et B, nous avons eu besoin de l’image centrée sur des espaces
« vides » pour reconstruire la totalité du plan (110).
Dans les directions AB les plus
proches, il n’y a aucune interaction. Des interactions se font en dehors des
positions atomiques hors directions AB les plus proches.
Cette répartition de densité de
charge est typique de Ge et elle est bien différente de celle de Si qui
concentre les accumulations de charge dans les plans (100).

Cette planche retrace le volume de la
densité de charge différence autour du point d’inversion χ en représentation
multipolaire.
Dans la représentation 3D, on note un
volume en forme de 2 trépieds inversés autour du point χ qui assure un réseau
continu dans l’espace 3D.

La planche 19 reste une
représentation multipolaire. Dans cette planche, nous avons associé volume 3D
et plans. Entre les plans (110) et (1─1 0), il est à remarquer l’inversion de
la figure suivant l’axe Z et plus généralement, il en est de même pour les axes
X et Y.


Ces planches montrent les densités
différences des vues pour une petite valeur de δρ. Les planches 20 et 21 concernent la
disposition de la partie interne des surfaces volumiques pour des rayons très
inférieurs aux rayons des domaines contenant la totalité de la charge de Si ou
Ge.
Notons que l’ondulation sur des
surfaces externes est due à l’obligation d’une fermeture informatique, elle a
pour valeur l’isodensité locale rencontrée, exemples : planches 5, 18,
19….
Le volume étudié est donc le volume
juste autour de la position atomique, voir figures 5 et 6 (4πr2ρ0(r)). Nous voyons que proches des
positions atomiques, les surfaces volumiques sont réparties suivant des axes
quaternaires hélicoïdaux facilement visibles pour les domaines haut et bas des
vues.
Pour la planche 20, il faut réaliser
que pour cette orientation 2 domaines de la densité électronique sont cachés et
qu’il y a 6 domaines d’isodensité 0.01 e─/ Å3 formant un
environnement géométrique de type octaédrique.
La planche 21 montre différentes
orientations des surfaces volumiques de la planche 20. L’ensemble des surfaces
volumiques observées est un environnement octaédrique près de la position
atomique.

Nous avons repris les domaines
volumiques contenant la totalité des électrons de Si (14 e─) et Ge
(32e─).
La structure globale est plus
compliquée. En particulier, la nature « noduleuse » de Si est bien
marquée. Ge garde la structure plus octaédrique comme vue précédemment.
Remarques
Les différences de couleur sur les
représentations 2D et 3D proviennent d’éclairages différents selon les
orientations. C’est un phénomène particulièrement visible lors des rotations
des plans. La position de la source lumineuse est fixe.
Les modèles multipolaires sont basés sur des hypothèses a priori et sur des
paramètres obtenus par moindres carrés c’est-à-dire obtenus en les considérant
tous à la fois dans un même affinement créant de fortes corrélations entre eux.
Cette méthode entraîne des solutions
multiples difficiles à évaluer quant à leur réalité.
De plus, dans le cas des modèles,
seule une représentation par cartes (2D) est souvent proposée sans faire une
correspondance avec les paramètres utilisés dans le modèle.
L’Analyse Directe
Multipolaire
Dans l’Analyse Directe Multipolaire,
la contribution électronique des atomes voisins est filtrée par la seule
présence dans la représentation mathématique des termes significatifs d'ordres
peu élevés du développement multipolaire de l'atome considéré. Ainsi, chaque
composante multipolaire peut être discutée séparément (PRESENTATION).
Il y a
une connaissance directe des couplages atomiques internes et externes. En même
temps, l’Analyse Directe Multipolaire
amoindrit le « bruit » généré, entre autres, par les
composantes non significatives et attire l’attention sur les seules propriétés
significatives. La majeure partie des propriétés physiques est due à
l’organisation de la répartition électronique. Dans le cas de la diffraction neutronique,
il s’agit de la distribution nucléaire.
L’Analyse Directe Multipolaire a des
avantages indéniables sur la représentation conventionnelle de Fourier. Il
devient possible de connaître l'origine des différents faits spatiaux en les
assignant à leurs atomes "parents" et de définir les types de
comportement multipolaire. L'information est alors découverte sous une forme
prête pour une interprétation immédiate.
Elle traite chaque paramètre
séparément ce qui évite les fortes
corrélations et permet de juger de la réalité de chaque paramètre.
Dès la représentation sous forme de
courbes (représentation 1D), l’association avec le comportement de chaque
harmonique sphérique donne la majorité des propriétés du composé (état ionique,
nombre d’électrons sous le pic de diffraction, directions des transferts de
charge,…).
Les autres représentations 2D (en
cartes) et 3D (visualisations) facilitent
la compréhension des phénomènes et donnent des vues plus faciles à
interpréter.
Mais l’essentiel de l’Analyse Directe
Multipolaire est inclus dans la représentation 1D.
L’avantage de la visualisation 3D est
une vue directe des surfaces d’isodensité soit dans la maille (Fourier
conventionnel), soit atome par atome (représentation multipolaire).
La comparaison de ces 2 types de
représentation permet de détecter les mécanismes des interactions entre les
atomes.
Comparaison entre Si et Ge
Dans le tableau des données de base,
il est à remarquer que la différence des paramètres de la maille cubique entre
Si et Ge est de 0,2336Å ce qui correspond à 4% d’augmentation pour Ge.
Par contre, la différence entre les
nombres d’électrons attribués à Si et Ge est de 18 e─ .
Les figures 5 et 6 montrent le
comportement de la composante zéro de Si et Ge. Ce sont les seules figures qui
ne sont pas en série différence grâce à la détermination du terme résiduel.
C’est une comparaison directe entre Fexp et Ftheor . On
peut noter la zone de densité relativement plate au-delà de 0,75Å. Dans les 2
cas Si et Ge, on ne détecte pas un comportement ionique ou métallique, ….
Les différences entre Si et Ge
interviennent à partir des figures 7 et 8 où l’inversion de la composante
d’ordre 3 de Si par rapport à Ge présente l’essentiel de la différence entre
les composantes de Si et Ge.
Nous retrouvons ces différences sur
les planches 1, 2, 3 relatives à l’Analyse Directe Multipolaire faite atome par
atome.
Pour Si, les illustrations Fourier conventionnel font ressortir le problème des
artefacts situés dans des zones de faible densité de charge. Ce phénomène n’est
visible que pour Si. La comparaison des cartes de Si et Ge soulignent bien que
ces maximums situés dans des espaces vides sont des artefacts (planches 7, 8,
10).
Pour Ge, la structure en zigzag dans les plans {110} est typique et relie les
sites A aux sites B dans la direction AB la plus grande. Dans la direction AB
la plus petite, nous avons au contraire un manque de densité de charge
(planches12, 13).
La planche 14 indique dans les plans
{001} une forte différence de comportement entre Si et Ge.
Les planches 20, 21 soulignent le
comportement octaédrique de la densité de charge différence proche du centre de
la position atomique.
En résumé, même si les positions atomiques respectent la même symétrie pour Si et
Ge, il est évident que les distributions de charge sont bien différentes.
Il n’est donc pas possible d’établir
sur la base des seules positions atomiques les propriétés physiques de composés
géométriquement analogues.
Chaque composé a ses propres
caractéristiques qui conduisent à des propriétés spécifiques.
Si et Ge en sont un exemple probant.
Etablir un modèle sans la
connaissance d’une analyse directe méticuleuse, approfondie, basée directement
sur les F(hkl) mesurés, est une méthode très hasardeuse.
C’est
par la connaissance expérimentale des lois phénoménologiques que les concepts
fondamentaux se précisent ou se modifient. La souplesse scientifique est de
confronter l’ensemble d’une théorie avec un ensemble de faits et d’adapter
progressivement les concepts fondamentaux de la Science.
Références
(1) K. KURKI-SUONIO
(1977) Isr. J. chem. 16, 115-129; 132-136
(2) M. KARA & K.
KURKI-SUONIO (1981) Acta Cryst. A37, 201-210
(3)
R.Saravanan, K.S. SYED ALI &
S. ISRAEL (2008) Pramana, J.
of physics, vol.70, n°4, 679-696
(4) R.W.
Alkire, W.B. Yelon and J. R. Schneider (1982) Phys. Rev. B26, 3097,
(5)
A. Kazimirov and V. G. Kohn
(2011) Acta Cryst. A67 409-414
(6) S.
Cummings & M. Hart (1988)
Aust.J. Phys., 41, 423-431,
(7) Z.
W. Lu, A. Zunger & M. Deutsch
(1993) Phys. Rew. B, 47, n°15,9385-9410
mag3D Figure1 Figure2 Figure3 PRESENTATION SYMPOSIUM FRANCO FINLANDAIS SYMPOSIUM Problème des phases The phase problem DVD Semi-conducteur organométallique organosilicié TiO2-rutile SiO2-stishovite Cu2O-cuprite K2PtCl6 Be-metal bases and 3D views Bases et vues 3D Si-et-Ge CaF2-fluorite NbC-2023